NEW DESIGN ALERT!This design comes from one of my favorite photos of mine of Gao Gao, a.k.a. "big big" who is small small, the panda powerhouse who fathered FIVE pandas during his tenure at the San Diego Zoo.
Go to Curryosities and get yourself some Small & Mighty swag and you can walk around proudly with a panda in your pocket! Get predesigned apparel or adjust where the pocket is to your liking by using the Customize tool! 
0 Comments
I do a lot of analyses in for loops that don’t inherently print anything to the terminal. Translation:  So, unless you echo some kind of progress statement in your script, there is zero indication of where it's at. Up until now, I would echo a progress statement but, if you have lots of samples, that can clutter the screen. What I wanted was something sleek, like a progress bar, but more informative than just a progress bar. Like a good Bioinformagician, I Googled and Googled trying to find what I wanted but, alas, I couldn't find anything simple that did what I wanted. So, I finally said enough is enough and spent a few days writing something that not only does exactly what I want but is also universal to any script with a for loop. I call it ThatLionLady's Loop-Progress. You can find all the nitty gritty details on how it works and how you can integrate it into your own scripts on my GitHub, but generally, this is how it works: Loop-Progress is an external script called within your own script (just like calling a program) that monitors the progress of your for loop through `if elif else` statements that determine what to print in the terminal based on the calculated percentage completed. As each loop completes, a new percentage is calculated and the previous progress output is overwritten on the terminal with the new progress output. This gives the appearance of a working progress bar! The information you get with each loop:
Now, if your for loop is using a program that has terminal output, it doesn’t mean you CAN’T use Loop-Progress (it does, afterall, provide so much useful information), it just means you’ll have to make a few minor changes to the script. For instance, removing the "n" flag from echo so the progress bar doesn’t overwrite that output making your screen look all weird. The same change can also be made to the script to keep all the output. Add a standard error log and you're now keeping track of how long each loop takes. This will now be a standard in my scripts and I hope, whether you poach pieces or use it as is, that this is helpful for your future for looping endevors!
I have opened an online shop where I’m selling all kinds of products with designs I've made from my photography.
If you haven't seen it on my Facebook or Instagram already, the link to my shop is curryosities.myspreadshop.com (or find it in the menu under Other Fun Stuff). This shop is dedicated to my dearly departed mutt, Paughey. He was the most adorably dopey good dog and looks pretty good on a t-shirt! My nephews wear their Paughey shirts proudly with Parker making sure those who comment on it know “he’s dead”. Kids say the darndest things!
I'll be releasing new designs periodically and introducing them here! So keep checking back until you find something you like. Or, if there's something you're interested in, a specific product or animal, LET ME KNOW! I'll do my best to make it happen! I’ve already done several custom orders to rave reviews.  While this was intended to be an artistic outlet, it’s also a side hustle of sorts, so I do get a portion of the profits from your purchase. Help a girl working for a non-profit, living with her parents out. This is a really niche post intended for geneticists who are doing de novo genome assemblies of 10X linked-reads sequencing data using Supernova (see… niche), but it also might be interesting to people who want to know a little bit about what the heck it is I do for a living. A good amount of bioinformatics is Googling.
Supernova is an assembly program specifically for use with linked-reads sequencing data generated by 10X Genomics sequencing technology. Explaining exactly what linked-reads are can be a whole other post so if you want those details, go here. But for now, all you need to know is that it’s the raw data I am using to generate reference genomes from scratch (a.k.a. de novo). The end product of a de novo assembly is something called a FASTA. This is a large file that contains all the As, Gs, Cs, and Ts in order for that genome. This file is what’s used to as a guide for doing future genomic analyses of the species. In Supernova, there are 4 options for this output. The online documentation didn't go into enough depth on how the 4 output options work for me to really know which would be the best option for what we’re going to be using them for, and Googling didn’t get me any closer to an answer. So, I generated all 4 types of output, got assembly stats (QUAST & BUSCO) on all of them, and that gave me enough info to figure out which option would be best. So, here's my very simplified takeaway from this endeavor: Supernova represents sequences in the raw assembly as “microbubbles” and “megabubbles”.  It looks like a “bubble” is when there's more than one sequence assembled to a contig separated by "gaps" (single sequences or runs of Ns). Collections of microbubbles create megabubbles. These megabubbles then have to be flattened into a single sequence. This is where the output options come into play. 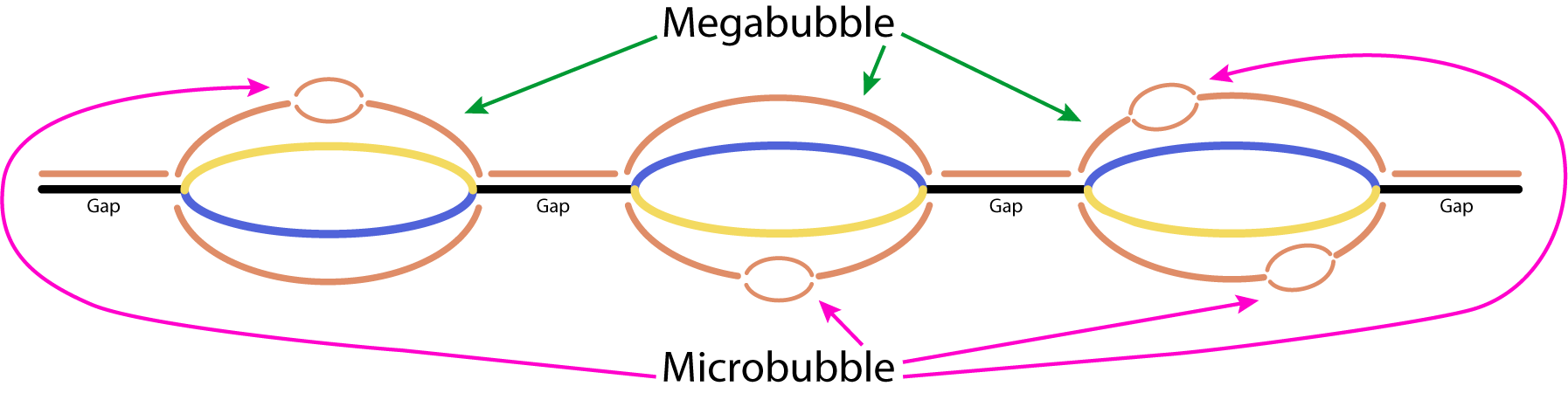 So, here’s the full breakdown that sounds like a riddle but isn’t a riddle: raw is ALL the bubbles even microbubbles within megabubbles in one FASTA megabubbles flattens microbubbles within megabubble arms in one FASTA pseudohap flattens all the megabubbles to one FASTA pseudohap2 is each flattened megabubble arm in a separate FASTA
I hope this helps some of my fellow Googling bioinfomagicians out there and didn’t make everyone else just super confused.  Find yourself a job that makes you well up with pride to be an employee every time you see new promotional material. San Diego Zoo Wildlife Alliance participated in the 133rd Rose Parade for the first time in 25 years. The float’s design represents the interconnectedness of all life with a live action depiction of our new past, present, and future in wildlife conservation logo and a rotating globe with all our Hubs.
|






